

On the left is a negatively-stained electron micrograph of a membrane vesicle isolated from the electric ray Torpedo california, with a muscle-type nicotinic single acetylcholine receptor (AcChR) pointed out . To the right the structure of the AcChR determined to NN resolution using cryoelectron microscopy by Rahman, Teng, Worrell, Noviello, Lee, Karlin, Stowell & Hibbs (2020). “Structure of the native muscle-type nicotinic receptor and inhibition by snake venom toxins.”
As a new assistant professor (1), I was called upon to teach my department’s “Cell Biology” course. I found, and still find, the prospect challenging in part because I am not exactly sure which aspects of cell biology are important for students to know, both in the context of the major, as well as their lives and subsequent careers. While it seems possible (at least to me) to lay out a coherent conceptual foundation for biology as a whole [see 1], cell biology can often appear to students as an un-unified hodge-podge of terms and disconnected cellular systems, topics too often experienced as a vocabulary lesson, rather than as a compelling narrative. As such, I am afraid that the typical cell biology course often re-enforces an all too common view of biology as a discipline, a view, while wrong in most possible ways, was summarized by the 19th/early 20th century physicist Ernest Rutherford as “All science is either physics or stamp collecting.” A key motivator for the biofundamentals project [2] has been to explore how to best dispel this prejudice, and how to more effectively present to students a coherent narrative and the key foundational observations and ideas by which to scientifically consider living systems, by any measure the most complex systems in the Universe, systems shaped, but not determined by, physical chemical properties and constraints, together with the historical vagaries of evolutionary processes on an ever-changing Earth.
Two types of information: There is an underlying dichotomy within biological systems: there is the hereditary information encoded in the sequence of nucleotides along double-stranded DNA molecules (genes and chromosomes). There is also the information inherent in the living system. The information in DNA is meaningful only in the context of the living cell, a reaction system that has been running without interruption since the origin of life. While these two systems are inextricably interconnected, there is a basic difference between them. Cellular systems are fragile, once dead there is no coming back. In contrast the information in DNA can survive death – it can move from cell to cell in the process of horizontal gene transfer. The Venter group has replaced the DNA of bacterial cells with synthetic genomes in an effort to define the minimal number of genes needed to support life, at least in a laboratory setting [see 3, 4]. In eukaryotes, cloning is carried out by replacing a cell’s DNA, with that of another cell (reference).
Conflating protein synthesis and folding with assembly and function: Much of the information stored in a cell’s DNA is used to encode the sequence of various amino acid polymers (polypeptides). While over-simplified [see 5], students are generally presented with the view that each gene encodes a particular protein through DNA-directed RNA synthesis (transcription) and RNA-directed polypeptide synthesis (translation). As the newly synthesized polypeptide emerges from the ribosomal tunnel, it begins to fold, and is released into the cytoplasm or inserted into or through a cellular membrane, where it often interacts with one or more other polypeptides to form a protein [see 6]. The assembled protein is either functional or becomes functional after association with various non-polypeptide co-factors or post-translational modifications. It is the functional aspect of proteins that is critical, but too often their assembly dynamics are overlooked in the presentation of gene expression/protein synthesis, which is really a combination of distinct processes.
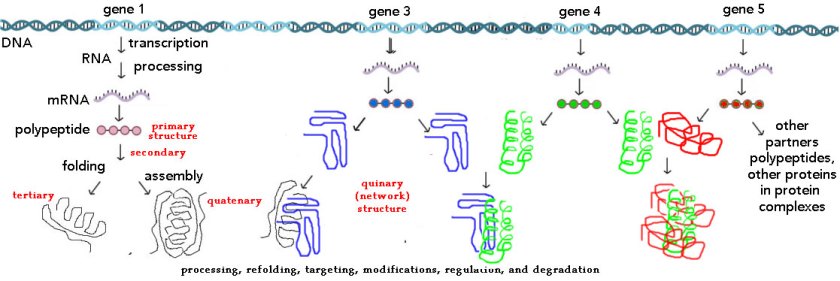
Students are generally introduced to protein synthesis through the terms primary, secondary, tertiary, and quaternary structure, an approach that can be confusing since many (most) polypeptides are not proteins and many proteins are parts of complex molecular machines [here is the original biofundamentals web page on proteins + a short video][see Teaching without a Textbook]. Consider the nuclear pore complex, a molecular machine that mediates the movement of molecules into and out of the nucleus. A nuclear pore is “composed of ∼500, mainly evolutionarily conserved, individual protein molecules that are collectively known as nucleoporins (Nups)” [7]. But what is the function of a particular NUP, particularly if it does not exist in significant numbers outside of a nuclear pore? Is a nuclear pore one protein? In contrast, the membrane bound, mitochondrial ATP synthase found in aerobic bacteria and eukaryotic mitochondria, is described as composed “of two functional domains, F1 and Fo. F1 comprises 5 different subunits (three α, three β, and one γ, δ and ε)” while “Fo contains subunits c, a, b, d, F6, OSCP and the accessory subunits e, f, g and A6L” [8]. Are these proteins or subunits? is the ATP synthase a protein or a protein complex?

Such confusions arise, at least in part, from the primary-quaternary view of protein structure, since the same terms are applied, generally without clarifying distinction, to both polypeptides and proteins. These terms emerged historically. The purification of a protein was based on its activity, which can only be measured for an intact protein. The primary structure of a polypeptide was based on the recognition that DNA-encoded amino acid polymers are unbranched, with a defined sequence of amino acid residues (see Sanger. The chemistry of insulin). The idea of a polypeptide’s secondary structure was based on the “important constraint that all six atoms of the amide (or peptide) group, which joins each amino acid residue to the next in the protein chain, lie in a single plane” [9], which led Pauling, Corey and Branson [10] to recognized the α-helix and β-sheet, as common structural motifs. When a protein is composed of a single polypeptide, the final folding pattern of the polypeptide, is referred to as its tertiary structure and is apparent in the first protein structure solved, that of myoglobin (↓), by Max Perutz and John Kendell.

Myoglobin’s role in O2 transport depends upon a non-polypeptide (prosthetic) heme group. So far so good, a gene encodes a polypeptide and as it folds a polypeptide becomes a protein – nice and simple (2). Complications arise from the observations that 1) many proteins are composed of multiple polypeptides, encoded for by one or more genes, and 2) some polypeptides are a part of different proteins. Hemoglobin, the second protein whose structure was

determined, illustrates the point (←). Hemoglobin is composed of four polypeptides encoded by distinct genes encoding α- and β-globin polypeptides. These polypeptides are related in structure, function, and evolutionary origins to myoglobin, as well as the cytoglobin and neuroglobin proteins (↓). In
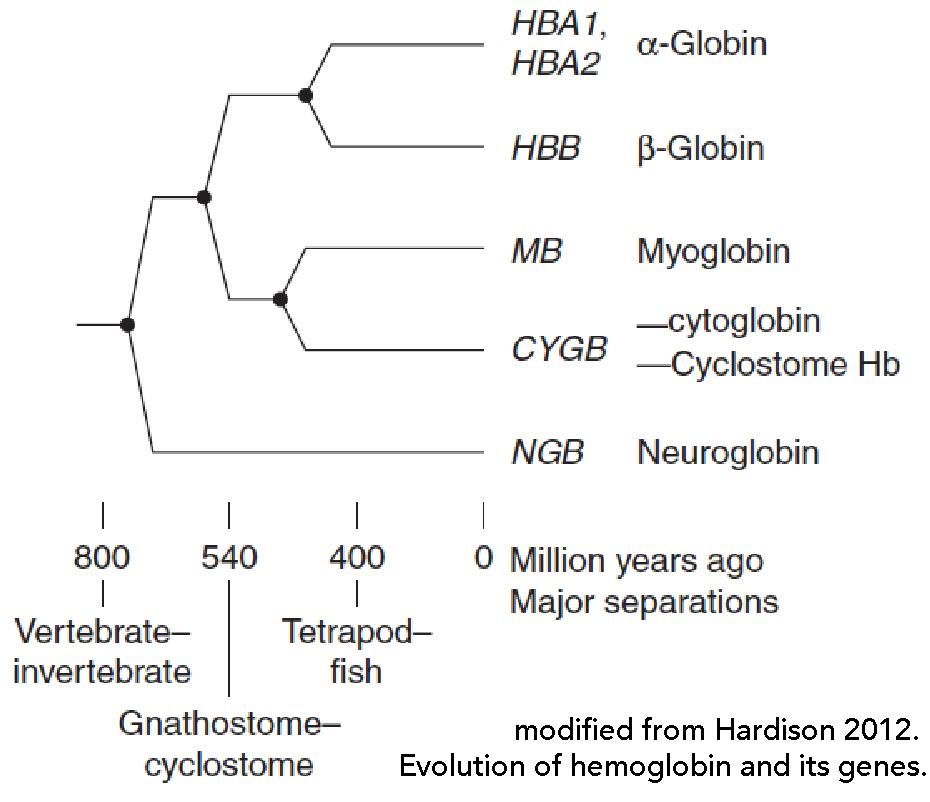
humans, there are a number of distinct α-like globin and β-like globin genes that are expressed in different hematopoetic tissues during development, so functional hemoglobin proteins can have a number of distinct (albeit similar) subunit compositions and distinct properties, such as their affinities for O2 [see 11].

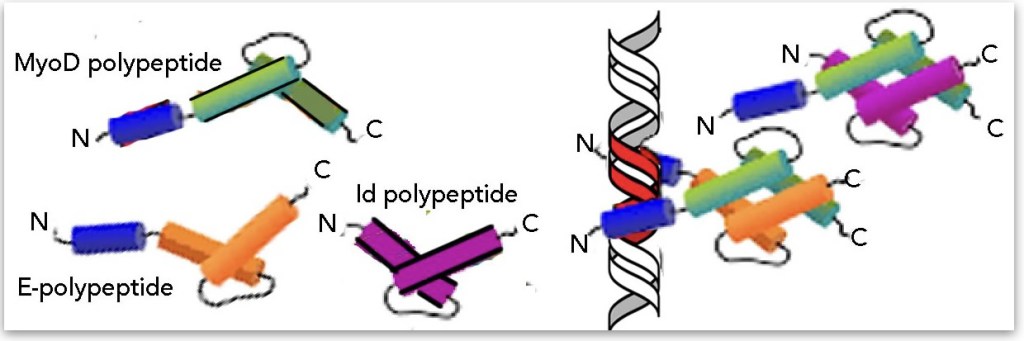
But the situation often gets more complicated. Consider centrin-2, a eukaryotic Ca2+ binding polypeptide that plays roles in organizing microtubules, building cilia, DNA repair, and gene expression [see 12 and references therein]. So, is the centrin-2 polypeptide just a polypeptide, a protein, or a part of a number of other proteins? As another example, consider the basic-helix-loop-helix family of transcription factors; these transcription factor proteins are typically homo- or hetero-dimeric; are these polypeptides proteins in their own right? The activity of these transcription factors is regulated in part by which binding partners they contain. bHLH polypeptides also interact with the Id polypeptide (or is it a protein); Id lacks a DNA binding domain so when it forms a dimer with a bHLH polypeptide it inhibits DNA binding (↓). So is a single bHLH polypeptide a protein or is the protein necessarily a dimer? More to the point, does the current primary→quaternary view of protein structure help or hinder student understanding of the realities of biological systems? A potentially interesting bio-education research question.
A recommendation or two: While under no illusion that the complexities of polypeptide synthesis and protein assembly can be easily resolved – it is surely possible to present them in a more coherent, consistent, and accessible manner. Here are a few suggestions that might provoke discussion. Let us first recognize that, for those genes that encode polypeptides: i) they encode polypeptides rather than functional proteins (a reality confused by the term “quaternary structure”). We might well distinguish a polypeptide from a protein based on the concentration of free monomeric polypeptide (gene product) within the cell. Then we need to convey the reality to students that the assembly of a protein is no simple process, particularly within the crowded cytoplasm [13], a misconception supported by the simple secondary-tertiary structure perspective. While some proteins assemble on their own, many (most?) cannot.

As an example, consider the protein tubulin (↑). As noted by Nithianantham et al [14], “ Five conserved tubulin cofactors/chaperones and the Arl2 GTPase regulate α- and β-tubulin assembly into heterodimers” and the “tubulin cofactors TBCD, TBCE, and Arl2, which together assemble a GTP-hydrolyzing tubulin chaperone critical for the biogenesis, maintenance, and degradation of soluble αβ-tubulin.” Without these various chaperones the tubulin protein cannot be formed. Here the distinction between protein and multiprotein complex is clear, since tubulin protein exists in readily detectable levels within the cell, in contrast to the α- and β-tubulin polypeptides, which are found complexed to the TBCB and TBCA chaperone polypeptides. Of course the balance between tubulin and tubulin polymers (microtubules) is itself regulated by a number of factors.
The situation is even more complex when we come to the ribosome and other structures, such as the nuclear pore. Woolford [15] estimates that “more than 350 protein and RNA molecules participate in yeast ribosome assembly, and many more in metazoa”; in addition to four ribsomal RNAs and ~80 polypeptides (often referred to as ribosomal proteins) that are synthesized in the cytoplasm and transported into the nucleus in association with various transport factors, these “assembly factors, including diverse RNA-binding proteins, endo- and exonucleases, RNA helicases, GTPases and ATPases. These assembly factors promote pre-rRNA folding and processing, remodeling of protein–protein and RNA–protein networks, nuclear export and quality control” [16]. While I suspect that some structural components of the ribosome and the nuclear pore may have functions as monomeric polypeptides, and so could be considered as proteins, at this point it is best (most accurate) to assume that they are polypeptides, components of proteins and larger, molecular machines (past post).
We can, of course, continue to consider the roles of common folding motifs, arising from the chemistry of the peptide bond and the environment within and around the assembling protein, in the context of protein structure [17, 18], The knottier problem is how to help students recognize how functional entities, proteins and molecular machines, together with the coupled reaction systems that drive them and the molecular interactions that regulate them, function. How mutations, alleleic variations, and various environmentally induced perturbations influence the behaviors of cells and organisms, and how they generate normal and pathogenic phenotypes. Such a view emphasizes the dynamics of the living state, and the complex flow of information out of DNA into networks of molecular machines and reaction systems.
Acknowledgements: Thanks to Michael Stowell for feedback and suggestions and Jon Van Blerkom for encouragement. All remaining errors are mine. Post updated to include imagines in the right places (and to include the cryoEM structure of the AcChR + minor edits – 16 December 2020.
Footnotes:
- Recently emerged from the labs of Martin Raff and Lee Rubin – Martin is one of the founding authors of the transformative “molecular biology of the cell” textbook.
- Or rather quite over-simplistic, as it ignore complexities arising from differential splicing, alternative promoters, and genes encoding non-polypeptide encoding RNAs.
Literature cited (please excuse excessive self-citation – trying to avoid self-plagarism)
1. Klymkowsky, M.W., Thinking about the conceptual foundations of the biological sciences. CBE Life Science Education, 2010. 9: p. 405-7.
2. Klymkowsky, M.W., J.D. Rentsch, E. Begovic, and M.M. Cooper, The design and transformation of Biofundamentals: a non-survey introductory evolutionary and molecular biology course. LSE Cell Biol Edu, in press., 2016. pii: ar70.
3. Gibson, D.G., J.I. Glass, C. Lartigue, V.N. Noskov, R.-Y. Chuang, M.A. Algire, G.A. Benders, M.G. Montague, L. Ma, and M.M. Moodie, Creation of a bacterial cell controlled by a chemically synthesized genome. science, 2010. 329(5987): p. 52-56.
4. Hutchison, C.A., R.-Y. Chuang, V.N. Noskov, N. Assad-Garcia, T.J. Deerinck, M.H. Ellisman, J. Gill, K. Kannan, B.J. Karas, and L. Ma, Design and synthesis of a minimal bacterial genome. Science, 2016. 351(6280): p. aad6253.
5. Samandi, S., A.V. Roy, V. Delcourt, J.-F. Lucier, J. Gagnon, M.C. Beaudoin, B. Vanderperre, M.-A. Breton, J. Motard, and J.-F. Jacques, Deep transcriptome annotation enables the discovery and functional characterization of cryptic small proteins. Elife, 2017. 6.
6. Hartl, F.U., A. Bracher, and M. Hayer-Hartl, Molecular chaperones in protein folding and proteostasis. Nature, 2011. 475(7356): p. 324.
7. Kabachinski, G. and T.U. Schwartz, The nuclear pore complex–structure and function at a glance. J Cell Sci, 2015. 128(3): p. 423-429.
8. Jonckheere, A.I., J.A. Smeitink, and R.J. Rodenburg, Mitochondrial ATP synthase: architecture, function and pathology. Journal of inherited metabolic disease, 2012. 35(2): p. 211-225.
9. Eisenberg, D., The discovery of the α-helix and β-sheet, the principal structural features of proteins. Proceedings of the National Academy of Sciences, 2003. 100(20): p. 11207-11210.
10. Pauling, L., R.B. Corey, and H.R. Branson, The structure of proteins: two hydrogen-bonded helical configurations of the polypeptide chain. Proceedings of the National Academy of Sciences, 1951. 37(4): p. 205-211.
11. Hardison, R.C., Evolution of hemoglobin and its genes. Cold Spring Harbor perspectives in medicine, 2012. 2(12): p. a011627.
12. Shi, J., Y. Zhou, T. Vonderfecht, M. Winey, and M.W. Klymkowsky, Centrin-2 (Cetn2) mediated regulation of FGF/FGFR gene expression in Xenopus. Scientific Reports, 2015. 5:10283.
13. Luby-Phelps, K., The physical chemistry of cytoplasm and its influence on cell function: an update. Molecular biology of the cell, 2013. 24(17): p. 2593-2596.
14. Nithianantham, S., S. Le, E. Seto, W. Jia, J. Leary, K.D. Corbett, J.K. Moore, and J. Al-Bassam, Tubulin cofactors and Arl2 are cage-like chaperones that regulate the soluble αβ-tubulin pool for microtubule dynamics. Elife, 2015. 4.
15. Woolford, J., Assembly of ribosomes in eukaryotes. RNA, 2015. 21(4): p. 766-768.
16. Peña, C., E. Hurt, and V.G. Panse, Eukaryotic ribosome assembly, transport and quality control. Nature Structural and Molecular Biology, 2017. 24(9): p. 689.
17. Dobson, C.M., Protein folding and misfolding. Nature, 2003. 426(6968): p. 884.
18. Schaeffer, R.D. and V. Daggett, Protein folds and protein folding. Protein Engineering, Design & Selection, 2010. 24(1-2): p. 11-19.

There is clearly a paradox with nomenclature in cell biology. In addition, knowing what is a protein and what is a protein complex is very difficult to decipher. Even with tubulin, alpha tubulin is a protein. Beta tubulin is a protein. But the dimer of the two is the basic functional unit. And if a protein can be defined as a functional unit, then the alpha/beta dimer should be the protein. But it is not.
I agree that the idea of primary to quaternary structure does not accomplish all we need. However, sometimes simple patterns and rules can be very helpful for learning. Somewhat like the idea of variations on a theme in music. Provide the theme, and then the variations can be better understood. With proteins, provide the theme of structure, and then the variations are more readily understood. But without knowledge of the basic pattern, the chaos of protein structure is too great.
Please note that this is only one of many problems in teaching cell biology. The terminology is horrible, and even the agreed-upon terms are less than helpful (ex: channels, transporters, pumps, cotransporters, or also GPCRs and enzyme-coupled Rs). We need to find some common ground that makes sense for students.
LikeLike