

Updates: 12 January 2022
-
Man Receives Transplanted Heart from Genetically Modified Pig
- First transplant of a genetically altered pig heart into a person sparks ethics questions
7 December 2020: US FDA declares genetically modified pork ‘safe to eat‘
A practical benefit, from a scientific and medical perspective, of the evolutionary unity of life (link) are the molecular and cellular similarities between different types of organisms. Even though humans and bacteria diverged more than 2 billion years ago (give or take), the molecular level conservation of key systems makes it possible for human insulin to be synthesized in and secreted by bacteria and pig-derived heart valves to be used to replace defective human heart valves (see link). Similarly, while mice, pigs, and people are clearly different from one another in important ways they have, essentially, all of the same body parts. Such underlying similarities raise interesting experimental and therapeutic possibilities.
A (now) classic way to study the phenotypic effects of human-specific versions of genes is to introduce these changes into a model organism, such as a mouse (for a review of human brain-specific human genes – see link). A example of such a study involves the gene that encodes the protein foxp2, a protein involved in the regulation of gene expression (a transcription factor). The human foxp2 protein differs from the foxp2 protein in other primates at two positions; 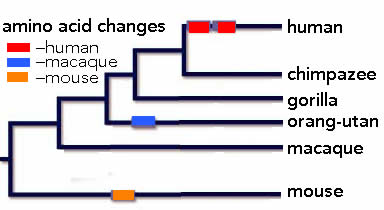 these two amio acid changes alter the activity of the human protein, that is the ensemble of genes that it regulates. That foxp2 has an important role in humans was revealed through studies of individuals in a family that displayed a severe language disorder linked to a mutation that disrupts the function of the foxp2 protein. Individuals carrying this mutant foxp2 allele display speech apraxia, a “severe impairment in the selection and sequencing of fine oral and facial movements, the ability to break up words into their constituent phonemes, and the production and comprehension of word inflections and syntax” (cited in Bae et al, 2015). Male mice that carry this foxp2 mutation display changes in the “song” that they sing to female mice (1), while mice carrying a humanized form of foxp2 display changes in “dopamine levels, dendrite morphology, gene expression and synaptic plasticity” in a subset of CNS neurons (2). While there are many differences between mice and humans, such studies suggest that changes in foxp2 played a role in human evolution, and human speech in particular.
these two amio acid changes alter the activity of the human protein, that is the ensemble of genes that it regulates. That foxp2 has an important role in humans was revealed through studies of individuals in a family that displayed a severe language disorder linked to a mutation that disrupts the function of the foxp2 protein. Individuals carrying this mutant foxp2 allele display speech apraxia, a “severe impairment in the selection and sequencing of fine oral and facial movements, the ability to break up words into their constituent phonemes, and the production and comprehension of word inflections and syntax” (cited in Bae et al, 2015). Male mice that carry this foxp2 mutation display changes in the “song” that they sing to female mice (1), while mice carrying a humanized form of foxp2 display changes in “dopamine levels, dendrite morphology, gene expression and synaptic plasticity” in a subset of CNS neurons (2). While there are many differences between mice and humans, such studies suggest that changes in foxp2 played a role in human evolution, and human speech in particular.

Another way to study the role of human genes using mouse as a model system is to generate what are known as chimeras, named after the creature in Greek mythology composed of parts of multiple organisms. A couple of years ago, Goldman and colleagues (3) reported that human glial progenitor cells could, when introduced into immune-compromised mice (to circumvent tissue rejection), displaced the mouse’s own glia, replacing them with human glia cells. Glial cells are the major non-neuronal component of the central nervous system. Once thought of as passive “support” cells, it is now clear that the two major types of glia, known as astrocytes and oligodendrocytes, play a number of important roles in neural functioning [back track post]. In their early studies, they found that the neurological defects associated with the shaker mutation, a mutation that disrupts the normal behavior of oligodendrocytes, could be rescued by the implantation of normal human glial progenitor cells (hGPCs)(4). Such studies confirmed what was already known, that the shaker mutation disrupts the normal function of myelin, the insulating structure around axons that dramatically speeds the rate at which neuronal signals (action potentials) move down the axons and activate the links between neurons (synapses). In the central nervous system, myelin is produced by oligodendrocytes as they ensheath neuronal axons. Human oligodendrocytes derived from hGPCs displaced the mouse’s mutation carrying oligodendrocytes and rescued the shaker mouse’s mutation-associated neurological defect.
Glial cells are the major non-neuronal component of the central nervous system. Once thought of as passive “support” cells, it is now clear that the two major types of glia, known as astrocytes and oligodendrocytes, play a number of important roles in neural functioning [back track post]. In their early studies, they found that the neurological defects associated with the shaker mutation, a mutation that disrupts the normal behavior of oligodendrocytes, could be rescued by the implantation of normal human glial progenitor cells (hGPCs)(4). Such studies confirmed what was already known, that the shaker mutation disrupts the normal function of myelin, the insulating structure around axons that dramatically speeds the rate at which neuronal signals (action potentials) move down the axons and activate the links between neurons (synapses). In the central nervous system, myelin is produced by oligodendrocytes as they ensheath neuronal axons. Human oligodendrocytes derived from hGPCs displaced the mouse’s mutation carrying oligodendrocytes and rescued the shaker mouse’s mutation-associated neurological defect.
 Subsequently, Goldman and associates used a variant of this approach to introduce hGPCs (derived from human embryonic stem cells) carrying either a normal or mutant version of the Huntingtin protein, a protein associated with the severe neural disease Huntington’s chorea (OMIM: 143100)(5). Their studies strongly support a model that locates defects associated with human Huntington’s disease to defects in glia. This same research group has generated hGPCs from patient-derived, induced pluripotent stem cells (patient-derived HiPSCs). In this case, the patients had been diagnosed with childhood-onset schizophrenia (SCZ) [link](6). Skin biopsies were taken from both normal and children diagnosed with SCZ; fibroblasts were isolated, and reprogrammed to form human iPSCs. These iPSCs were treated so that they formed hGPCs that were then injected into mice to generate chimeric (human glial/mouse neuronal) animals. The authors reported systematic differences in the effects of control and SCZ-derived hGPCs; “SCZ glial mice showed reduced prepulse inhibition and abnormal behavior, including excessive anxiety, antisocial traits, and disturbed sleep”, a result that suggests that defects in glial behavior underlie some aspects of the human SCZ phenotype.
Subsequently, Goldman and associates used a variant of this approach to introduce hGPCs (derived from human embryonic stem cells) carrying either a normal or mutant version of the Huntingtin protein, a protein associated with the severe neural disease Huntington’s chorea (OMIM: 143100)(5). Their studies strongly support a model that locates defects associated with human Huntington’s disease to defects in glia. This same research group has generated hGPCs from patient-derived, induced pluripotent stem cells (patient-derived HiPSCs). In this case, the patients had been diagnosed with childhood-onset schizophrenia (SCZ) [link](6). Skin biopsies were taken from both normal and children diagnosed with SCZ; fibroblasts were isolated, and reprogrammed to form human iPSCs. These iPSCs were treated so that they formed hGPCs that were then injected into mice to generate chimeric (human glial/mouse neuronal) animals. The authors reported systematic differences in the effects of control and SCZ-derived hGPCs; “SCZ glial mice showed reduced prepulse inhibition and abnormal behavior, including excessive anxiety, antisocial traits, and disturbed sleep”, a result that suggests that defects in glial behavior underlie some aspects of the human SCZ phenotype.
The use of human glia chimeric mice provides a powerful research tool for examining the molecular and cellular bases for a subset of human neurological disorders. Does it raise a question of making mice more human? Not for me, but perhaps I do not appreciate the more subtle philosophical and ethical issues involved. The mice are still clearly mice, most of their nervous systems are composed of mouse cells, and the overall morphology, size, composition, and organization of their central nervous systems are mouse-derived and mouse-like. The situation becomes rather more complex and potentially therapeutically useful when one talks about generating different types of chimeric animals or of using newly developed genetic engineering tools (the CRISPR CAS9 system found in prokaryotes), that greatly simplify and improve the specificity of the targeted manipulation of specific genes (link). In these studies the animal of choice is not mice, but pigs – which because of their larger size produce organs for transplantion that are similar in size to the organs of people (see link). While similar in size, there are two issues that complicate pig to human organ transplantation: first there is the human immune system mediated rejection of foreign tissue and second there is the possibility that transplantation of porcine organs will lead to the infection of the human recipient with porcine retroviruses.
The issue of rejection (pig into human), always a serious problem, is further exacerbated by the presence in pigs of a gene encoding the enzyme α-1,3 galactosyl transferase (GGTA1). GGTA1 catalyzes the addition of the gal-epitope to a number of cell surface proteins. The gal-epitope is “expressed on the tissues of all mammals except humans and subhuman primates, which have antibodies against the epitope” (7). The result is that pig organs provoke an extremely strong immune (rejection) response in humans. The obvious technical fix to this (and related problems) is to remove the gal-epitope from pig cells by deleting the GGTA1 enzyme (see 8). It is worth noting that “organs from genetically engineered animals have enjoyed markedly improved survivals in non-human primates” (see Sachs & Gall, 2009).
 The second obstacle to pig → human transplantation is the presence of retroviruses within the pig genome. All vertebrate genomes, including those of humans, contain many inserted retroviruses; almost 50% of the human genome is retrovirus-derived sequence (an example of unintelligent design if ever there was one). Most of these endogenous retroviruses are “under control” and are normally benign (see 9). The concern, however, is that the retroviruses present in pig cells could be activated when introduced into humans. To remove (or minimize) this possibility, Niu et al set out to use the CRISPR CAS9 system to delete these porcine endogenous retroviral sequences (PERVs) from the pig genome; they appear to have succeeded, generating a number of genetically modified pigs without PERVs (see 10). The hope is that organs generated from PERV-minus pigs from which antigen-generating genes, such as α-1,3 galactosyl transferase, have also been removed or inactivated together with more sophisticated inhibitors of tissue rejection, will lead to an essentially unlimited supply of pig organs that can be used for heart and other organ transplantation (see 11), and so alleviate the delays in transplantation, and so avoid deaths in sick people and the often brutal and criminal harvesting of organs carried out in some countries.
The second obstacle to pig → human transplantation is the presence of retroviruses within the pig genome. All vertebrate genomes, including those of humans, contain many inserted retroviruses; almost 50% of the human genome is retrovirus-derived sequence (an example of unintelligent design if ever there was one). Most of these endogenous retroviruses are “under control” and are normally benign (see 9). The concern, however, is that the retroviruses present in pig cells could be activated when introduced into humans. To remove (or minimize) this possibility, Niu et al set out to use the CRISPR CAS9 system to delete these porcine endogenous retroviral sequences (PERVs) from the pig genome; they appear to have succeeded, generating a number of genetically modified pigs without PERVs (see 10). The hope is that organs generated from PERV-minus pigs from which antigen-generating genes, such as α-1,3 galactosyl transferase, have also been removed or inactivated together with more sophisticated inhibitors of tissue rejection, will lead to an essentially unlimited supply of pig organs that can be used for heart and other organ transplantation (see 11), and so alleviate the delays in transplantation, and so avoid deaths in sick people and the often brutal and criminal harvesting of organs carried out in some countries.
The final strategy being explored is to use genetically modified hosts and patient derived iPSCs to generate fully patient compatible human organs. To date, pilot studies have been carried out, apparently successfully, using rat embryos with mouse stem cells (see 12 and 13), with much more preliminary studies using pig embryos and human iPSCs (see 14). The approach involves what is known as chimeric embryos. In this case, host animals are genetically modified so that they cannot generate the organ of choice. Typically this is done by mutating a key gene that encodes a transcription factor directly involved in formation of the organ; embryos missing pancreas, kidney, heart,  or eyes can be generated. In an embryo that cannot make these organs, which can be a lethal defect, the introduction of stem cells from an animal that can form these organs can lead to the formation of an organ composed primarily of cells derived from the transplanted (human) cells.
or eyes can be generated. In an embryo that cannot make these organs, which can be a lethal defect, the introduction of stem cells from an animal that can form these organs can lead to the formation of an organ composed primarily of cells derived from the transplanted (human) cells.
At this point the strategy appears to work reasonably well for mouse-rat chimeras, which are much more closely related, evolutionarily, than are humans and pigs. Early studies on pig-human chimeras appear to be dramatically less efficient. At this point, Jun Wu has been reported as saying of human-pig chimeras that “we estimate [each had] about one in 100,000 human cells” (see 15), with the rest being pig cells. The bottom line appears to be that there are many technical hurdles to over-come before this method of developing patient-compatible human organs becomes feasible. Closer to reality are PERV-free/gal-antigen free pig-derived, human compatible organs. The reception of such life-saving organs by the general public, not to mention religious and philosophical groups that reject the consumption of animals in general, or pigs in particular, remains to be seen.
figures reinserted & minor edits 23 October 2020 – new link 17 December 2020.
references cited
- A Foxp2 Mutation Implicated in Human Speech Deficits Alters Sequencing of Ultrasonic Vocalizations in Adult Male Mice.
- A Humanized Version of Foxp2 Affects Cortico-Basal Ganglia Circuits in Mice
- Modeling cognition and disease using human glial chimeric mice.
- Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination.
- Human glia can both induce and rescue aspects of disease phenotype in Huntington disease
- Human iPSC Glial Mouse Chimeras Reveal Glial Contributions to Schizophrenia.
- The potential advantages of transplanting organs from pig to man: A transplant Surgeon’s view
- see Sachs and Gall. 2009. Genetic manipulation in pigs. and Fisher et al., 2016. Efficient production of multi-modified pigs for xenotransplantation by ‘combineering’, gene stacking and gene editing
- Hurst & Magiokins. 2017. Epigenetic Control of Human Endogenous Retrovirus Expression: Focus on Regulation of Long-Terminal Repeats (LTRs)
- Nui et al., 2017. Inactivation of porcine endogenous retrovirus in pigs using CRISPR-Cas9
- Zhang 2017. Genetically Engineering Pigs to Grow Organs for People
- Kobayashi et al., 2010. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells.
- Kobayashi et al., 2015. Targeted organ generation using Mixl1-inducible mouse pluripotent stem cells in blastocyst complementation.
- Wu et al., 2017. Interspecies Chimerism with Mammalian Pluripotent Stem Cells
- Human-Pig Hybrid Created in the Lab—Here Are the Facts

One thought on “Humanized mice & porcinized people”